Install Seq2Fun
Seq2Fun was written in C/C++11 and can be installed on Linux or Mac OS X (with Xcode and Xcode Command Line Tools installed). We have bench marked Seq2Fun on Ubuntu (16.04 LTS and above) and macOS Catalina equipped with Intel and AMD CPU types. It may not work well on ARM-based CPU (used in Apple M1 chip and Tau VM)
Download & compile Click here to check and download all Seq2Fun stable releases (including version 1). The latest development version is hosted on GitHub. After download, from within the folder containing the downloaded package, issue the following commands:
>tar -xvzf seq2fun_v*.tar.gz
>cd Seq2Fun/src/
>make clean
>make
or the latest version, please follow instruction in Seq2Fun github
-
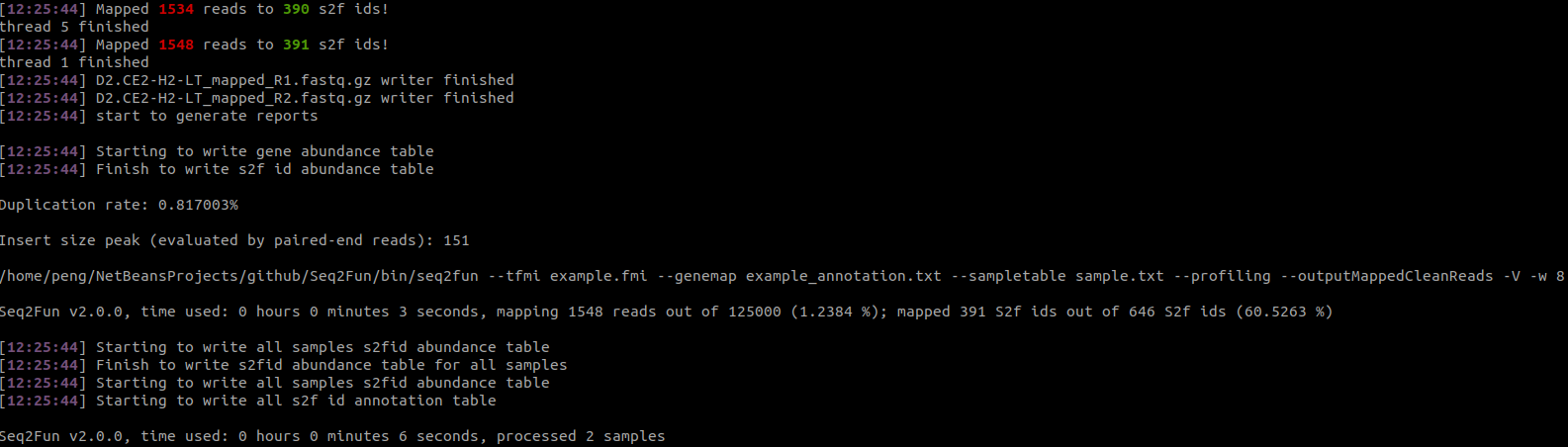
Run a small test There are four sub folders under seq2fun - src, bin, database and testdata. The bin folder contains the binary code we just complied. The testdata contains a small test data from the Case Study. Please issue the following commands:
>cd ../testdata/
>../bin/seq2fun --sampletable sample.txt --tfmi example.fmi --genemap example_annotation.txt -w 8 --profiling -V --outputMappedCleanReads --outputReadsAnnoMapThe progress will be displayed on the terminal. The screenshot below (from Ubuntu 20.04.3 LTS) shows the last few lines:
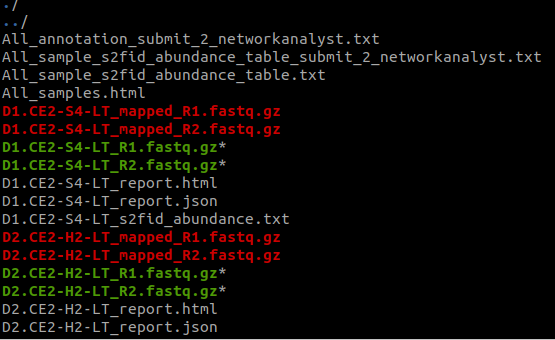
You will see many files generated in the current folder:
-
Submit gene abundance table to our website-based tool ExpressAnalyst (optional) for comprehensive data analysis and visual exploration. Seq2Fun generates several main files of s2f gene abundance table
S2fid_abundance_table_all_samples.txt, S2fid_abundance_table_all_samples_submit_2_expressanalyst.txt, the last one can be used to submit to ExpressAnalyst.
Note: please do not submit the tables generated by the testdata to ExpressAnalyst for the downstream analysis. This abundance table will not generate any biologically meaningful results because it is heavily subsampled of the original data.
Please go to ExpressAnalyst and select the example data DC cormorant toxicity for testing.