How does Seq2Fun work?
The Seq2Fun workflow has three main functions:
- Raw reads quality control
- Translated search in a protein database
- Abundance table generation
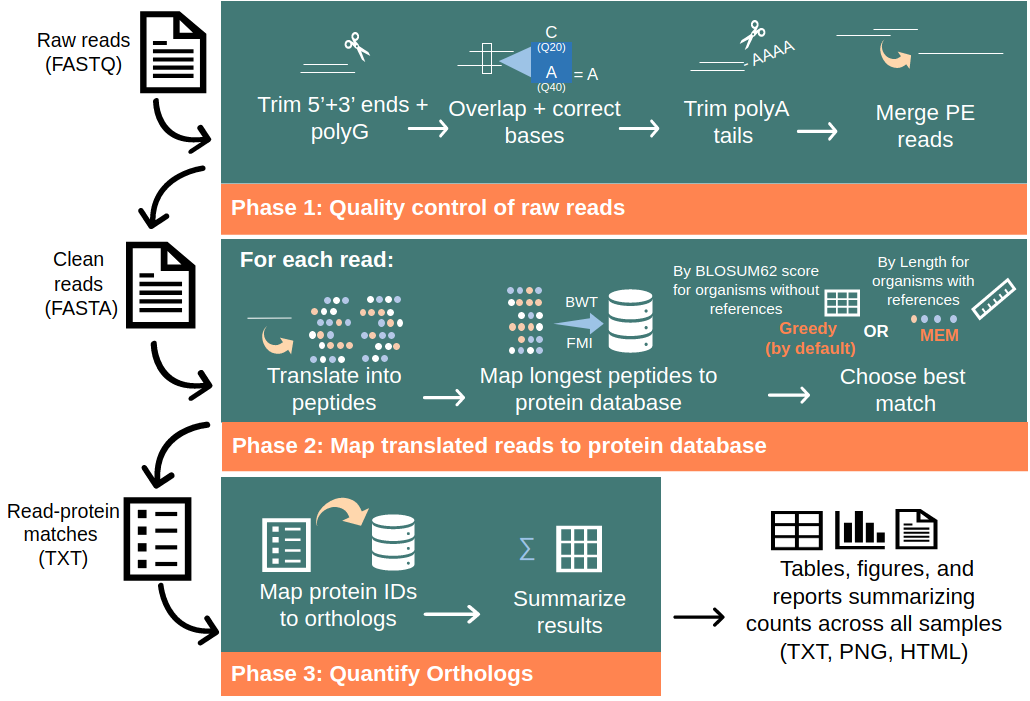
1. Quality control of raw RNA-seq reads
Seq2Fun loads reads in packs (n = 1000 reads). Each read is trimmed at both 5' and 3' ends, followed by removing polyG tail, which has been a common issue for Illumina NextSeq and NovaSeq. Low complexity sequences are also removed because they could cause artificially high protein matching score. If reads are paired-end (PE), overlapping region will be analyzed for error correction. Next, sequencing adopters and ployA tail are also removed before merging the overlapped read pair to form clean reads.
2. Translated search
The translated search (DNA-to-protein BLATX search) with high speed is the core algorithm of any modern searching tool to cope with the explosion of sequencing data.
Full-text index in Minute space (FM) index based on Burrow Wheeler transformation (BWT) is one of the modern fast pattern searching data structures.
BWT compresses and produces a reversible transformation of a block of reference sequences, which save storage space.
The FM method constructs a data structure based on combination of BWT compressed data and suffix arrays.
It uses the BWT to generate efficient indices and guarantees fast search with time linear to the length of the search string without decompressing the entire sequence.
It also sets various checkpoints and thus keeps the memory footprint small.
Therefore, Seq2Fun adopted FM index, which enables fast and sensitive mapping.
Each clean read is translated into all possible amino acid (AA) fragments using all reading frames, which could result in dozens of aa fragments with various lengths. But only the longest AA fragments are sent for homologies search in the protein database consisting of ortholog sequences.
For a typical mature mRNA, it consists of a 5’ cap, 5’ untranslation region (UTR), coding region (CDS), 3’ UTR and ployA tail.
For the clean reads, reads containing ployA tails have already been removed in reads quality check step.
The typical lengths of 5’ and 3’ UTRs range from 100 to 200 nt, and 200 to 700 nt, respectively.
Reads from UTRs could be still translated but highly possibly produce uninformative matching.
On the other hand, with the increasing reads length and size of library inserts, the chances for PE reads overlapped with CDS will steadily be increased.
The majority of reads from mRNA libraries should originate from CDS and the only true peptide should be among the longest translated aa fragments without a stop codon in most cases.
Therefore, only at most the top six longest peptide fragments (although it could be more AA fragments for Seq2Fun Greedy mode) from the unmerged reads (e.g. for 100 bp reads, at most four fragments of 33 and two fragments of 32 aa from six fragments will be kept,
and one of them must be the true protein fragment) are kept for any further downstream analysis.
It is possible that, this could remove some true protein fragments which are from reads that contains either true start or stop codons,
however, this would have minimal effects on gene abundance counting as well as gene fold-changes.
In some cases, only keeping the top longest fragments could filter out a proportion of fragments that originated from the merged PE reads if start and stop codons are present in the middle of the reads.
Therefore, we recap the maximum cut-off length of peptide fragment to be 60 aa (by default, user can change this cut-off),
which will prevent the filtering out of some true peptide fragments from the merged reads.
In addition, for most cases, keeping the top longest peptide fragments should have very limited impact on the overall analysis especially for comparison purpose.
Seq2Fun employs two modes maximum exact match (MEM) and Greedy modes for translated search.
For the MEM mode, all the peptide fragments are sorted by length, and searching starts from the longest one with a minimum required match length m with the reference sequences.
This m mainly governs the trade-off between sensitivity and precision.
It is designed for organisms with reference genomes in a database.
However, for most non-model organisms, there are usually little to no reference resources available.
Using the MEM model could cause rapid decline in sensitivity with the increasing evolutionary distances and results in a large number of unmapped reads.
Therefore, the Greedy mode is introduced to cope with this challenge.
The Greedy mode first sorts peptide fragments based on the amino acid substitution model BLOSUM62 scores and searching is initiated from the fragment with highest score.
It subsequently performs exact matching with a seed length of 7 and extends matching at the left end while allowing substitutions (by default 2) until it reaches the end of the query or the maximum number of mismatches is reached.
If a fragment with the highest score has a hit and if it has the highest matching score than any other fragment, the search is terminated.
The matching protein id is assigned to this query read if the matching score is above the threshold score.

Only search the longest AA fragments. The translation generates dozens of AA fragments, but only one (highlighted in green) is the true AA fragment, which is also among the longest fragments. Therefore, Seq2Fun only picks up the longest fragments, although it could be more AA fragments with different BLOSUM62 scores, for homologies search in protein database.
3. Abundance table generation
First, cases where reads were mapped to multiple protein IDs are dealt with. Most often, these proteins are homologies from same or different organisms and share the same ortholog ID. If this is not the case, the ortholog with the highest frequency is used. After ensuring that each read is matched to a single ortholog ID, the final quantification is a simple summation of all read-ortholog matches. An additional html report is generated to report the summary of these tables in figure and table formats, plus a rarefaction curve with sequence depth against number of mapped orthologs for each sample, which can be used to determine the minimum informative number of reads for each sample. Importantly, Seq2Fun provides an option to choose number of reads to process.