Welcome to the Seq2Fun!
Seq2Fun is an ultrafast, all-in-one functional profiling tool for RNA-seq data analysis for organisms without reference genomes.
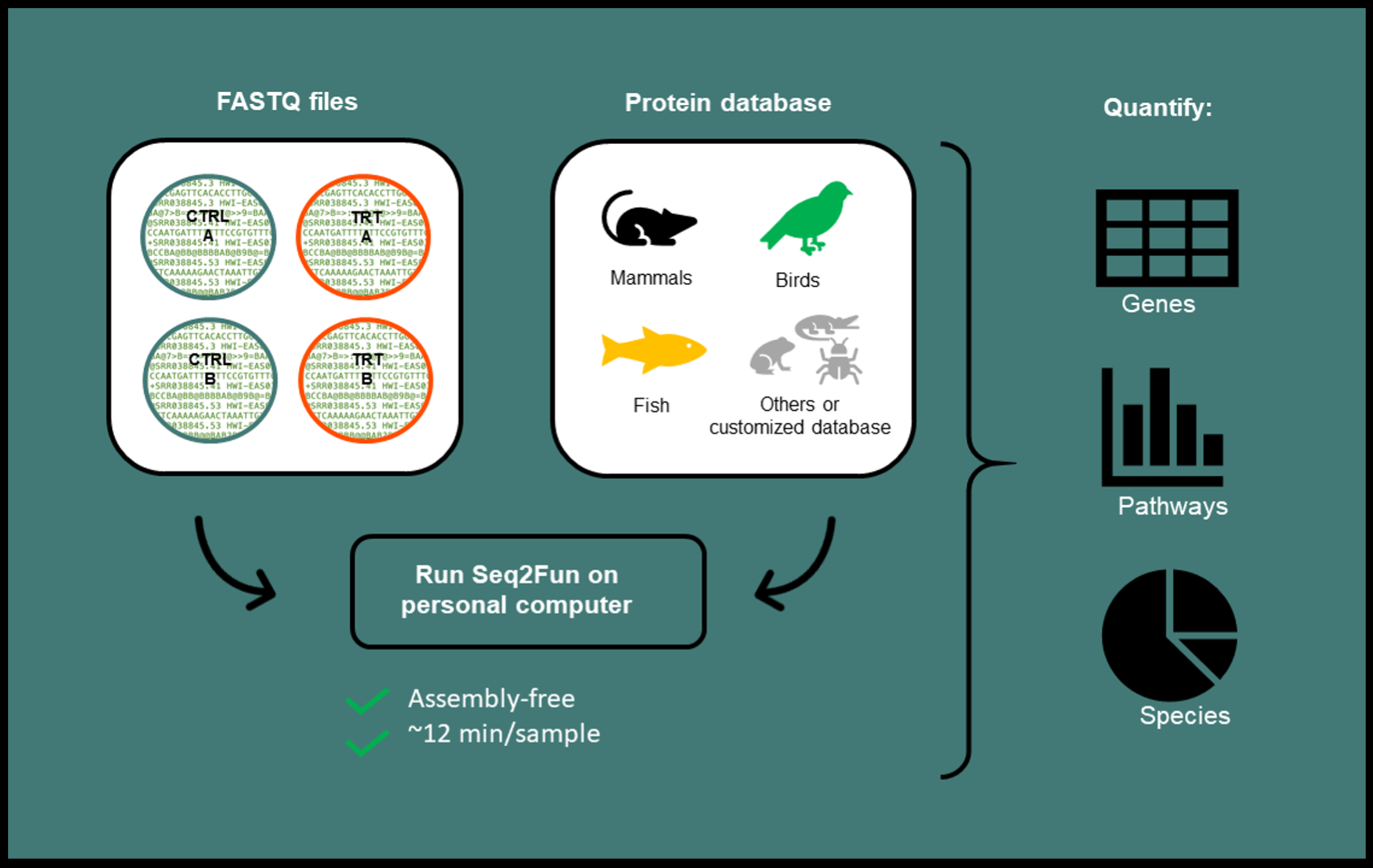
Key features of Seq2Fun
- Ultra-fast: > 120 times faster (~ 2 million reads / minute) than the conventional RNA-seq workflow.
- Extremely low memory cost: consumes as little as 2.27 GB memory and can run on a standard PC with 8 threads and 16 GB memory.
- Highly efficient: finish a typical RNA-seq dataset within several hours on a standard PC in stead of several days or even weeks by conventional RNA-seq workflow on a high-performance server.
- Reference-free: does not require the genome or transcriptome reference of the organism; it is also transcriptome de novo assembly-free.
- Highly accurate: generates KO abundance with R2 value as high as 0.93 comparing with the ground truth.
- High coverage: conducts gene quantification covering all the protein-coding genes.
- High resolution: generates gene abundance table at ortholog level.
- Multiple functional summaries: summarizes gene functional at ortholog, KO and GO level.
- All-in-one: directly takes raw RNA-seq reads as input and output gene abundance table without any intermediate file writing and loading, making I/O very efficient.
- Multifunctional: generates several levels of output files, including ortholog abundance table, a html report summarizing these tables and reads quality check, as well as output mapped clean reads for further analysis such as gene assembly.
- Target gene assemble: supports mapped reads extraction and de novo assemble for the target genes.
- Highly flexible: supports RNA-seq analysis on particular genes or groups of organisms using customized database.
- Easy to use: requires minimal programing skills.
News and Updates
- 02-17-2023 - migrate databases to expressanalyst.
- 08-29-2022 - seq2fun_v2.0.5 released.
- 08-01-2022 - seq2fun_v2.0.4 released.
- 07-26-2022 - updated databases based on phylogenetic analysis.
- 03-06-2022 - changed networkanalyst to expressanalyst for downstream analysis and update core orthologs.
- 03-02-2022 - added more prebuilt databases
- 02-27-2022 - seq2fun_v2.0.3 released (added core ortholog genes to the abundant table)
- 01-27-2022 - seq2fun_v2.0.2 released (removal of empty columns in output files)
- 12-22-2022 - seq2fun_v2.0.0 released (supporting high-resolution mapping with ortholog database)
- 08-30-2021 - seq2fun_v1.2.5 released
- 08-23-2021 - seq2fun_v1.2.4 released
- 06-18-2021 - seq2fun_v1.2.3 released
- 05-06-2021 - seq2fun_v1.2.2 released
- 03-31-2021 - seq2fun_v1.2.1 released
- 03-26-2021 - seq2fun_v1.2.0 released
- 03-22-2021 - seq2fun_v1.1.4 released
- 01-14-2021 - seq2fun_v1.1.2 released
- 12-06-2020 - seq2fun_v1.1.0 released
- 08-24-2020 - seq2fun_v1.0.0 released
Click here to check all the versions.
Citing Seq2Fun
Peng Liu, Jessica Ewald, Zhiqiang Pang, Elena Legrand, Yeon Seon Jeon, Jonathan Sangiovanni, Orcun Hacariz, Guangyan Zhou, Jessica A. Head, Niladri Basu, Jianguo Xia. (2023) “ExpressAnalyst: A unified platform for RNA-sequencing analysis in non-model species” Nature Communications (https://doi.org/10.1038/s41467-023-38785-y)
Peng Liu, Jessica Ewald, Jose Hector Galvez, Jessica Head, Doug Crump, Guillaume Bourque, Niladri Basu, Jianguo Xia. (2021) “Ultrafast functional profiling of RNA-seq data for nonmodel organisms” Genome Research (10.1101/gr.269894.120)